NEBNext®RNA 去除核心试剂 收藏
Download:
- isoschizomers |
- compatible ends |
- single letter code
相关产品:
NEBNext® RNA 去除核心试剂 – 含 RNA 纯化磁珠
产品特点:
RNAse-H 的高效工作流程结合在线工具设计的紧密间距探针,无论 RNA 质量高低,无论 RNA 起始量多少,都能高效去除 rRNA
· ·使用 NEBNext® Custom RNA Depletion Design Tool 设计的探针序列,可从任意物种中去除不需要的 RNA
· ·起始量范围广:10 ng – 1 μg
· · 同时适用于低质量和高质量 RNA 样本
· · 超快速工作流程:仅需 2 小时,手动操作时间少于 10 分钟
· · 可提供 RNAClean® 磁珠
概述:
在 RNA-seq 中,高丰度转录本如核糖体 RNA(rRNA)会占据绝大部分测序数据,但它的生物学意义极低,并会掩盖那些具有真正生物学意义的低丰度转录本的检测。当某种样品没有相应的 RNA 去除试剂盒时,这一挑战变的更为严峻。NEBNext® RNA 去除核心试剂提供客户定制,可以从任意物种中去除不需要的 RNA,探针设计工具操作简单方便(NEBNext® Custom RNA Depletion Design Tool)。
工作流程:
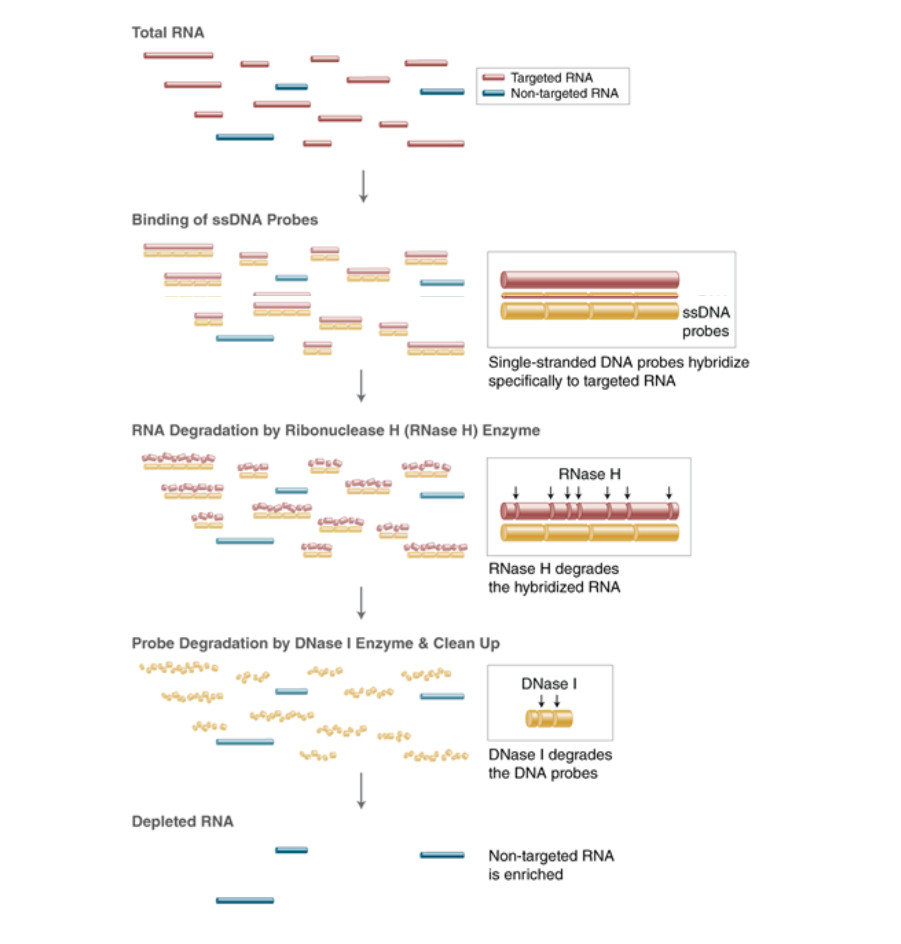
通过联合使用 NEB 提供的 NEBNext® RNA 去除核心试剂和客户自行设计定制的探针,去除不需要的 RNA,工作流程如下:
第一步:使用在线的 NEBNext® Custom RNA Depletion Design Tool,输入目标 RNA 序列,获得定制的探针序列。
第二步:从您信任的供应商处订购 ssDNA 寡核苷酸探针。
第三步:探针与 NEBNext® 定制 RNA 去除核心试剂一起使用,或与其它 NEBNext® RNA 去除试剂盒联合使用。
产品描述:
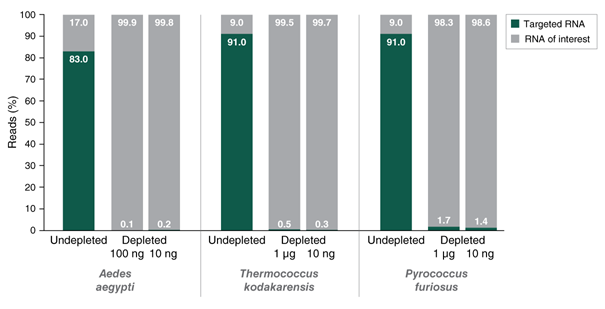
不同起始量、不同物种的样本,NEBNext® 定制 RNA 去除试剂盒均能高效去除目标 RNA,富集目的 RNAs。使用 NEBNext® Custom RNA Depletion Design Tool 针对埃及伊蚊、超嗜热原始菌和激烈火球菌的 rRNA 设计探针。使用 RNA 去除核心试剂盒和设计的探针从 1 µg、100 ng 和 10 ng 总 RNA 中去除 rRNA。使用 NEBNext® Ultra™ II RNA 定向文库制备试剂盒制备文库,并进行 Illumina 双端测序(2 x 75 bp)。从每个文库中抽取(seqtk)2000 万个 Reads。使用 Mirabait(6 个或更多,25-mers)鉴定 rRNA,残留的 rRNA 水平是通过将匹配上的 Reads 数除以质控过滤后的总 Reads 数计算得出。数据代表 3 次重复的平均值。结果表明:不同起始量(1 µg – 10 ng)、不同物种的样本,NEBNext® 定制 RNA 去除方法均能高效去除目标 RNA。
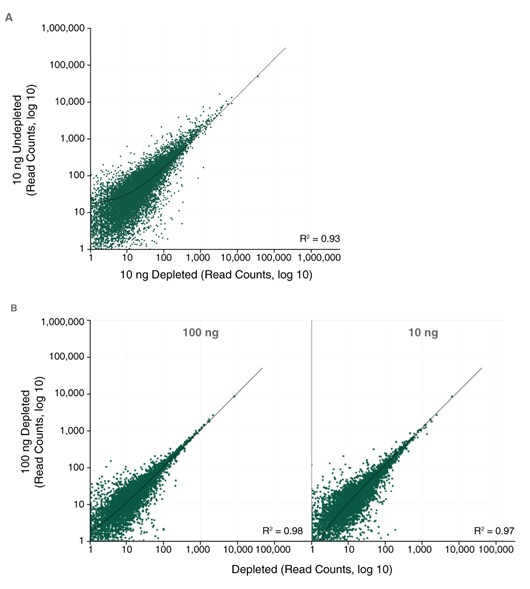
使用 NEBNext® 定制 RNA 去除试剂盒去除目标 RNA,不会影响目的转录本的丰度。使用 NEBNext® Custom RNA Depletion Design Tool 针对埃及伊蚊的 rRNA 设计探针。埃及伊蚊成虫(Benzon Research)。使用 Monarch® 总 RNA 小量提取试剂盒(NEB #T2010S)从埃及伊蚊成虫(Benzon Research)中提取总 RNA。使用 RNA 去除核心试剂盒和设计的探针从 100 ng 和 10 ng 总 RNA 中去除 rRNA。使用 NEBNext® Ultra™ II RNA 定向文库制备试剂盒制备文库,并进行 Illumina 双端测序(2 x 75 bp)。从去除后的文库中抽取(seqtk)2000 万个 Reads,从未去除的文库中抽取 2 亿个 Reads。使用 Salmon 和来自 Vectorbase(AaegL5.2 assembly)的转录本计算转录本的丰度。图中显示了线性拟合的 Read 计数和 R2 值。图 A 表明:去除不影响目的转录本的丰度。图 B 表明:不同起始量在多次重复实验中转录本丰度保持一致。
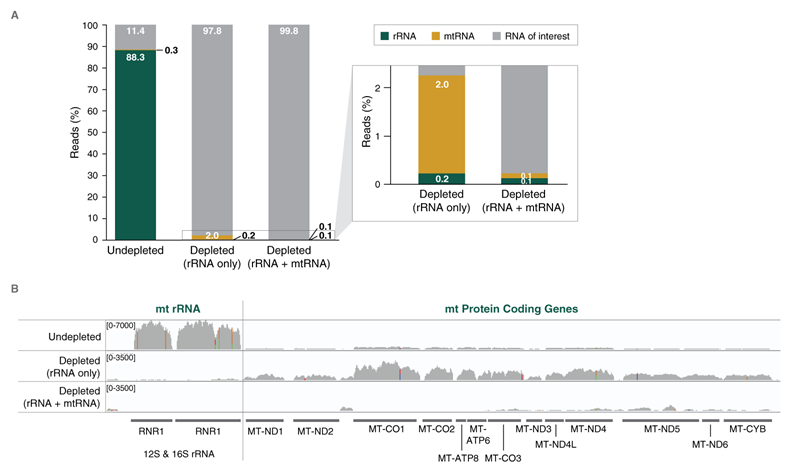
组合探针能有效去除人 rRNA 和线粒体 mRNA。使用 NEBNext® Custom RNA Depletion Design Tool 针对人线粒体 mRNA 设计探针。设计的探针与 NEBNext® rRNA 去除试剂盒 v2(人/小鼠/大鼠)的探针组合使用。使用组合探针从 1 µg 人通用参考总 RNA(Agilent®)中去除线粒体 RNA 和 rRNA。使用 NEBNext® Ultra™ II RNA 定向文库制备试剂盒制备文库,并进行 Illumina 双端测序(2 x 75 bp)。从每个文库中抽取(seqtk)2000 万个 Reads。图(A)使用 Mirabait(6 个或更多,25-mers)鉴定 rRNA 和线粒体 RNA,残留的 rRNA 和 mtRNA 水平是通过将匹配上的 Reads 数除以质控过滤后的总 Reads 数计算得出。rRNA 和线粒体 RNA 都被有效地去除。图 B 中 IGV 图显示了人类线粒体基因上的覆盖度。
实验流程: